

As the new medical devices Regulation (EU) 2017/745 became fully applicable on May 26th, 2021, medical device manufacturers are legally required to ensure that procedures, systems, documentation related to medical devices are in compliance.
The Medical Device Regulation (MDR) retain the existing principles and fundamental components of EU Medical Device Directive (MDD) but with significant improvements in each element. These improvements allowing effective, consistent, and robust regulatory framework for medical devices across EU Member states.
Some key improvements include:
New definition of medical device: the definition of MDs extended to include non-medical devices (No clinical benefit) for cleaning and sterilising devices and cosmetic devices such as coloured contact lenses, fillers, and liposuction devices.
Medical device classification: the new EU MDR 2017/745, Article 51, classified medical devices (MDs) into I, IIa, IIb, and III, considering their intended purposes and their inherent risks. The risk level increases from Class I to Class III, which means that the risk classification of MDs will have a direct impact on CE marking and regulatory obligations.
Incident reporting: all medical device manufacturers within EU member states are strictly required to report any serious and unexpected adverse incident that happens and Field Safety Corrective Action (FSCAs) to EU’s Competent Authorities. The time period for reporting serious and unexpected incidents have reduced from 30 to 15 days. Any serious incident that happens outside of EU Member states and that did not lead to FSCA do not need to be reported through vigilance in EU.
Periodic reports: all medical devices marketed in EU member states require either a Post Market Surveillance Report (PMSR) or PSUR. The risk classification of MDs determines type of report required.
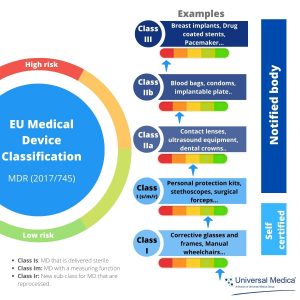
- Class I MDs require a PMSR available to the competent authority – recommended to be updated every 2-5 years.
- Class IIa require a PSUR available to notified body and, upon request, to competent authorities– update, when necessary, at least every 2 years.
- Class IIb and III require the submission of a PSUR – update at least once a year.
Technical documentation: the manufacturers are required to have an updated technical documentation for all classes of medical devices and accessible for competent authorities and notified bodies whenever required. Moreover, manufacturer required to establish, implement, document, and maintain a risk management system for each medical device (per MD or type of MD).
EUDAMED Reporting: as the launch of EUDAMED is delayed until May 2022, manufacturers should continue to report incidents directly to the national competent authority/authorities.
Contact our expert today to find out more about the new EU Medical Device regulation and how you can successfully implement it in your company:
Email – contact@universalmedica.com
Phone – 33(01) 41 12 27 77