

As we approach the end of the IVDR transition period, after May 26, 2022, all new in vitro diagnostic devices and Class A non-sterile IVD devices will need to be IVDR compliant in order to be marketed in the European Union. This means that all manufacturers of CE-marked IVD devices will be affected by the new regulation and any IVD devices that do not meet the new requirements will no longer be able to enter the European market.
Due to COVID’s backlog, the European Commission has approved the extension of transitional deadlines for products already on the EU market before May 26, 2022, to ensure continuity of supply for vital products.
IVDR Transition timelines
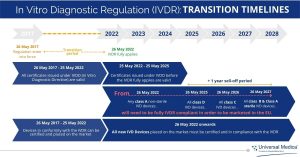
The new In Vitro Diagnostic Regulation aims to ensure consistent and high levels of patient safety throughout the EU member states. The IVDR has the same basic requirements as the previous regulation, IVDD, but with some important improvements to modernize the current system.
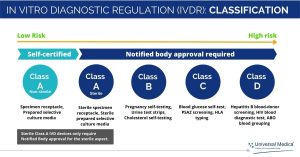
Compared to the IVDD, the IVDR is more rigorous, particularly with respect to risk classes and surveillance by Notified Bodies. This means that most IVD devices that currently only require self-certification will soon need to involve a Notified Body as part of the conformity assessment process.
According to the new regulation, IVD devices are classified into Class A, B, C or D, depending on the intended use of the device and its inherent risks!
As the transition period to IVDR draws to a close, it is important for in vitro diagnostic device manufacturers to stay on top of the changes and ensure compliance. Contact our experts today to learn how Universal Medica can help you navigate the complexities of the IVDR transition.
Sources