

Comme le nouveau règlement (UE) 2017/745 sur les dispositifs médicaux est devenu pleinement applicable le 26 mai 2021, les fabricants de dispositifs médicaux sont légalement tenus de s’assurer que les procédures, les systèmes et la documentation liés aux dispositifs médicaux sont conformes.
Le règlement sur les dispositifs médicaux (RDM ou MDR – Medical Device Regulation) conserve les principes existants et les composants fondamentaux de la directive européenne sur les dispositifs médicaux (DDM ou MDD – Medical Device Directive) avec des améliorations significatives dans chaque élément. Ces améliorations permettent un cadre réglementaire efficace, cohérent et solide pour les dispositifs médicaux dans les États membres de l’UE.
Certaines améliorations clés incluent :
Nouvelle définition du dispositif médical : la définition des DM a été étendue pour inclure les dispositifs non médicaux (sans avantage clinique) tels que les dispositifs de nettoyage et de stérilisation et les dispositifs cosmétiques tels que les lentilles de contact colorées, les produits de comblement et les dispositifs de liposuccion.
Classification des dispositifs médicaux : l’article 51 du nouveau règlement européen 2017/745 classe les dispositifs médicaux (DM) en I, IIa, IIb et III, compte tenu de leurs objectifs et de leurs risques inhérents. Le niveau de risque augmentant de la classe I à la classe III, la classification des risques des DM a un impact direct sur le marquage CE et les obligations réglementaires.
Signalement d’incident : tous les fabricants de dispositifs médicaux des États membres de l’UE sont strictement tenus de signaler tout incident indésirable grave et inattendu qui se produit et toute action corrective de sécurité sur le terrain (FSCA – Field Safety Corrective Action) aux autorités compétentes de l’UE. Le délai de signalement des incidents graves et inattendus est passé de 30 à 15 jours. Tout incident grave qui se produit en dehors des États membres de l’UE et qui n’a pas conduit à une FSCA n’a pas besoin d’être signalé par la vigilance dans l’UE.
Rapports périodiques : tous les dispositifs médicaux commercialisés dans les États membres de l’UE nécessitent soit un rapport de surveillance après commercialisation ou un rapport périodique actualisé de sécurité (PSUR). La classification des risques des DM détermine le type de rapport requis.
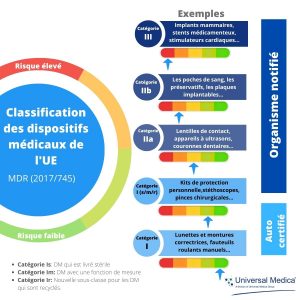
- Les DM de classe I nécessitent la mise à disposition aux Autorités de Santé d’un Rapport de Surveillance après commercialisation – il est recommandé de le mettre à jour tous les 2 à 5 ans.
- La classe IIa nécessite un PSUR mit à disposition à l’Organisme Notifié en charge de l’évaluation de la conformité et sur demande des Autorités Compétentes– mise à jour, si nécessaire, au moins tous les 2 ans.
- Les classes IIb et III nécessitent la soumission d’un PSUR – mise à jour au moins une fois par an.
Documentation technique : les fabricants sont tenus de disposer d’une documentation technique à jour pour toutes les classes de dispositifs médicaux et accessible aux autorités compétentes et aux organismes notifiés chaque fois que cela est nécessaire. De plus, le fabricant est tenu d’établir, de mettre en œuvre, de documenter et de maintenir un système de gestion des risques pour chaque dispositif médical (par DM ou type de DM).
Notification EUDAMED : le lancement d’EUDAMED ayant été retardé jusqu’en mai 2022, les fabricants doivent continuer à signaler les incidents directement aux autorités nationales compétentes.
Contactez notre expert dès aujourd’hui pour en savoir plus sur la nouvelle réglementation européenne sur les dispositifs médicaux et sur la manière dont vous pouvez la mettre en œuvre avec succès dans votre entreprise :
Courriel – contact@universalmedica.com
Téléphone – 33(01) 41 12 27 77